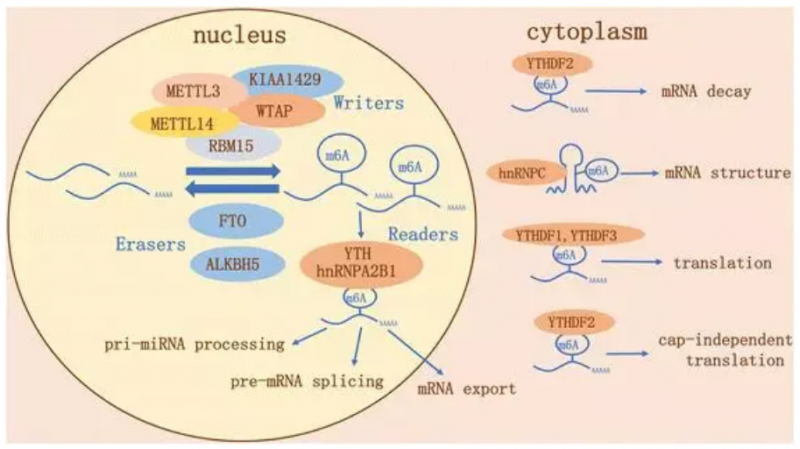
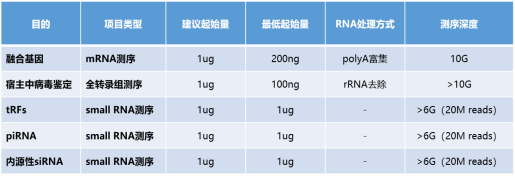
Q: 什么是 Visium 空间转录组?
Visium 空间转录组是在组织原位检测全转录组基因表达的一种技术,使得我们在检测基因表达水平的同时,获得基因在组织内部空间表达的位置信息。与空间转录组相比,传统的全基因转录组或单细胞转录组测序,丢掉了基因或细胞在组织内的空间分布信息,而广泛应用的 RNA 探针杂交,这只能同时检测有限的几个基因在组织内的空间表达分布。而空间转录组可以将基因表达与 H &E 染色的结果叠加在一起,不需要对组织进行消化,从而保留了组织内部的空间结构,这样使得我们可以研究组织内部不同区域的细胞异质性。
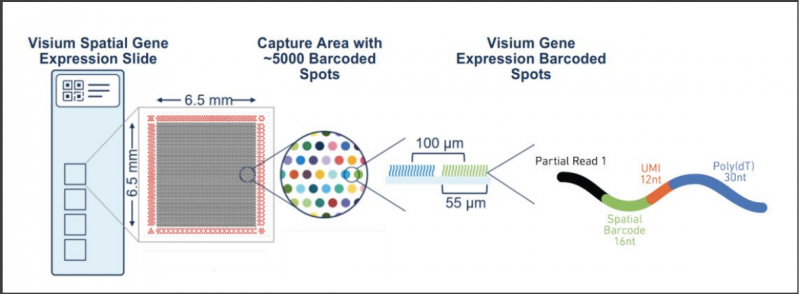
Visium 基因表达玻片上有 4 个捕获区域,也就是图片上展示的 4 个小方框,每个捕获区域的大小是 6.5mm 乘 6.5mm,每个捕获区域大约有 5000 个 spot,也就是图上所示的小圆点,每个 spot 的直径是 55µm,两个 spot 的中心点的距离是 100µm,每个 spot 上面有数百万条 oligo。每个 spot 上所有 oligo 的 Spatial Barcode 的序列都是相同的,而不同的 spot 之间 Spatial Barcode 是不同的,这样通过 Spatial Barcode,就能够知道每条 mRNA 到底位于哪个 spot,也就获得了 mRNA 的空间位置信息。UMI 用于基因表达定量,可以知道每个 spot 上有哪些基因表达以及他们的表达量。poly- A 用于捕获 mRNA。4 个捕获区域内的 Spatial Barcode 是相同的,但每个捕获区域内不同的 spot 之间 Spatial Barcode 是不同的。
总实验流程分为三个部分:
一是样本制备及预实验,二是组织优化,三是基因表达。
首先获取组织样本,进行冷冻,OCT 包埋,将包埋好的组织进行冰冻切片,将切好的组织放置于常规玻片上。接下来对组织切片进行固定,H& E 染色,用带扫描功能的显微镜对组织切片进行明场拍照。同时切 10 片厚度为 10 µm 的冰冻切片,提取 RNA,通过计算 RIN 值来评估其质量。为了获得先进实验结果,建议用 RIN 值大于 7 的组织块做 Visium 的实验。
首先对 OCT 包埋的组织进行冰冻切片,将切好的组织放置于组织优化玻片上的捕获区域。接下来对组织切片进行固定,H&E 染色,用带扫描功能的显微镜对组织切片拍照,拍照完成后会继续对组织切片进行通透处理,使细胞内的 mRNA 释放出来,被玻片上的 oligo 所捕获,在合成一链 cDNA 的时候,掺入带有荧光基团的碱基,这样合成好的 cDNA 就带有荧光,接下来会通过酶消化去除玻片上残留的组织,暴露带有荧光的 cDNA,用 Cy3 通道的荧光显微镜,扫描整张片子,获得荧光染色的图片,组织优化的所有实验都在玻片上完成。
先对 OCT 包埋的组织进行冷冻切片,将切好的组织切片放置于基因表达玻片上的捕获区域。接下来对组织切片进行固定,H&E 染色,用带扫描功能的显微镜对组织切片进行拍照,获得组织形态信息。拍照完成后会继续对组织切片进行通透处理,使细胞内的 mRNA 释放出来,被玻片上的 oligo 所捕获,在玻片上进行一链和二链 cDNA 的合成,合成二链 cDNA 后会做变性处理,将合成好的二链 cDNA 回收到 ep 管中,后面的所有实验都在 ep 管中完成,包括 cDNA 的扩增和文库的构建。空间基因表达实验可以大致分为两部分,首要部分从组织固定到二链 cDNA 的合成,都是在基因表达玻片上完成。第二部分从 cDNA 的扩增到文库的构建,都是在 ep 管中完成。
之所以用异戊烷冻存组织,而不是将组织直接放置于液氮中,是因为液氮的沸点比较低,如果将组织直接放到液氮里,液氮会沸腾,在组织周围形成气穴,导致组织不同区域的降温速度不同,容易在速冻过程中改变组织内部的形态,甚至使组织发生碎裂。而异戊烷的沸点比较高,将组织放置于异戊烷中,异戊烷不会沸腾,这样组织不同区域降温速度相同,就避免了在速冻过程中发生变形或者是碎裂。
目前只有包埋在 OCT 中的新鲜冷冻组织经过了 Visium 空间基因表达解决方案的验证。10X Genomics 的研发团队成员正在积极研究 FFPE 组织相关的应用,并且已经获得一些初步结果,预计在 2021 年会推出适用于 FFPE 组织的实验方案。
目前,Visium 空间基因表达流程是针对 H &E 染色而优化的。将 IHC 染色整合到此流程中是 10X Genomics 研发团队积极研究的另一个领域,预计在 2020 年上半年推出。
不会,10X Genomics 官方实验结果表明,H&E 染色不仅不会对样本 RNA 质量产生影响,染色后还会提高基因检测数。
运输组织样本:先用异戊烷冻存,将冻存好的组织放在密封的容器中,再将装有冷冻组织的容器用密封袋封好,放置于干冰中运输,样本到达目的地后迅速转移到 -80℃冰箱保存。
运输已经贴了组织切片的玻片:可以将玻片放置于密封的容器中,并将玻片固定好,再将装有玻片的容器用密封的袋子封好,放置于干冰中运输。同样样本到达目的地后,迅速转移到 -80℃冰箱进行保存。这里还要注意,一旦玻片上贴好了组织切片,只能保存 7 天。
在 10X 测试过的组织类型中,有三种组织还需要进一步的优化,包括样本制备的优化和组织通透性的优化,分别是皮肤,胰腺和骨组织。
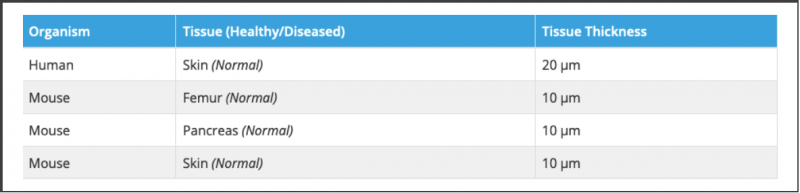
10X Genomics 测试过很多组织类型,大多数都可以用于空间转录组的分析。下图是他们官网上测试过的组织类型的列表,测试过的组织都可以运用在空间转录组的分析上面。但建议用户用基因表达玻片做正式实验前,务必用组织优化的玻片测试一下,看组织能否用于空间转录组的实验,同时也找到好的组织通透时间。
切片厚度基本上以 10µm 为主,但也有少量组织例外。

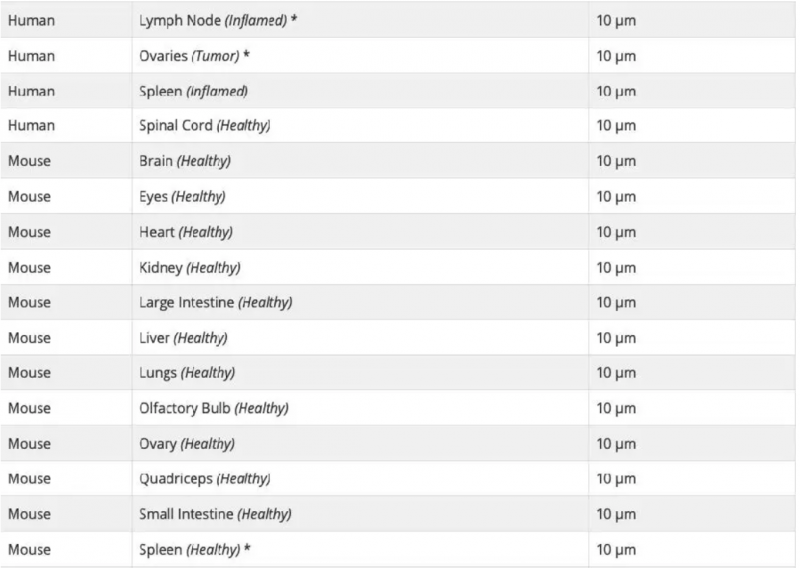
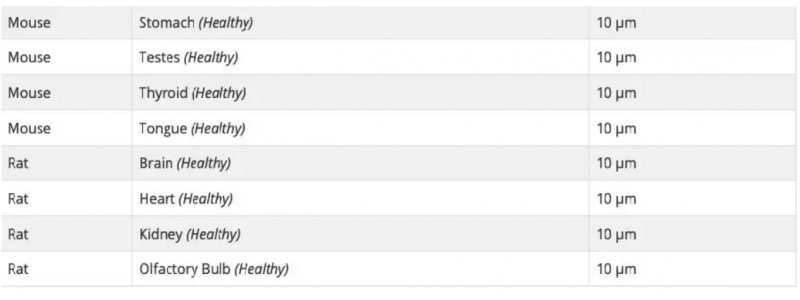
可以,Visium 空间基因表达玻片上可以检测不同类型的组织。每张玻片包含 4 个捕获区域,因此一张玻片上至多可检测四种不同类型组织的切片。每种组织好的透化时间需要通过组织优化(TO)实验来预先确定,每种类型的组织使用一张 TO 玻片。
是的,根据组织中待研究的具体区域,带条形码的捕获点可能覆盖不止一种类型的细胞。具体取决于组织类型、组织内的细胞大小以及这些细胞的密度。研究人员可看到每个点大约捕获 1 到 10 个细胞。您可以将每个捕获点想象为一种“微型批量”的实验,在这个实验中,研究人员收集一小批细胞的平均基因表达数据。这是 10x Genomics 的研究团队正在积极研究的另一领域,目的是尽量让捕获点变得更小。
无论您在处理过程中是否使用了所有捕获区域,玻片仅供一次性使用。这是因为流程中包含了多个步骤,包括对玻片进行染色、冲洗和重新干燥,这些步骤会破坏未使用的捕获区域上的寡核苷酸条形码,并降低其整体的灵敏度。
Visium 配件试剂盒中的成像测试玻片有两个用途。一,确定客户的成像系统是否与 Visium 兼容。二,用于成像程序的设置,以便在 Visium 组织优化和基因表达流程中获取图像。
在捕获区域四周有一圈由灰色小点组成的基准边界,提供组织切片的空间定位信息。
Visium 空间转录组的文库,可以在 Illumina 所有测序仪上测序,文库的结构如下图所示,需要双端 index 测序。
建议的测序方式是为 Read 1:28 个 bp,i7 index:10 个 bp,i5 index:10 个 bp,Read 2:120bp。Read 1 用于测试 Spatial Barcode 和 UMI,Read 2 用于测试插入片段。不建议将 visium 的文库与其他单 index 的 10x 文库混合测序。

与单细胞实验不同,我们无法根据细胞数计算空间转录组文库的测序量,因为我们不知道组织切片当中有多少细胞,我们根据组织切片占捕获区域的比例计算测序量,计算测序量的时候,只考虑被组织切片覆盖的 spot,每个 spot 的推荐底限起始测序量是 5 万个 read pairs,根据 H &E 染色的结果可以估算捕获区域到底有多少百分比的 spot 被组织覆盖。
计算测序量很简单,比如我们假设一个组织覆盖了捕获区 50% 的面积,我们就用 0.5×5000×5 万个 read pairs per spot,得出测序量是 125 个 million read pairs。这里面 0.5 就代表着覆盖了捕获区域 50% 的面积,5000 就代表着每一个捕获区域总共有 5000 个 spot,再乘以 5 万,5 万就是每个 spot 我们推荐的底限测序的量,就是 5 万个 read pairs per spot。测序量与组织大小、组织类型和基因表达水平相关。
通常,研究人员需要具备一点点生物信息学经验来使用 Space Range。Space Range 流程在 Linux 服务器上运行;这就需要研究人员了解基本的命令行操作,以及如何登录和退出 Linux 服务器。不过,在大多数情况下,Space Range 是高度自动化的。让软件正常运作的文件和算法都已经包含在内。因此,研究人员只需在测序后将两个输入文件提供给 Space Range,让软件生成空间基因表达数据。
Loupe Browser 有着完全图形化的界面,并且根本不需要编程。不过,您在研究过程中可能会碰到某个时刻,比如想了解空间基因表达数据的一个具体问题,或者创建一个自定义图形。那么,这就需要您学习 R 语言或其他脚本编程语言。
抱歉,不可以。因为数据类型不同,它们无法兼容。如果您有这种类型的数据,仍然建议您使用现有的处理空间转录组学数据的分析流程。
没错!事实上,分析空间基因表达数据的第三方工具的数量正在迅速增长。目前的一些选择包括 Spaniel 和 Giotto。Seurat 也是一种选择。Space Range 流程的输出矩阵与 Cell Ranger 的 Feature Barcode 输出矩阵的格式相同。研究人员可将此输出文件直接输入 Seurat 中进行进一步的分析。您可以通过 https://satijalab.org/seurat/v3.1/spatial_vignette.html 了解 Seurat 流程处理空间基因表达数据的更多信息。
此外,10x Genomics 的计算生物学家还创建了 R 资源,可以帮助客户对其样本开展深度分析。这些 R 资源让研究人员能够一次观察多个基因、样本或特征,如基因、UMI 和聚类。如果研究人员想分析不同特征的样本,或不同实验条件下的样本(如野生型、患病和治疗中的样本),这可能特别有用。您可以在我支持网站上访问这些 R 资源(https://support.10xgenomics.com/spatial-gene-expression/software/pipelines/latest/rkit)。